Spektrenbeschreibung
Durch welche Deskriptoren können Strukturen und Spektren einheitlich beschrieben werden?
Die Beschreibung von Spektren durch einen Code konstanter Länge ist verhältnismäßig einfach, da moderne Spektrometer die Spektren bereits in codierter Form, als eine Reihe von Werten, die bestimmten Wellenzahlen zugeordnet werden, ausgibt.
Gebräuchliche Größen (Deskriptoren) sind Transmission T und Absorption E , die wie folgt zusammenhängen:
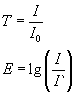
Zusammenhang von Transmission und Absorption
Datenreduktion
Spektrendaten können in der Form, wie sie durch das Spektrometer ausgegeben werden, zu Korrelationsuntersuchungen eingesetzt werden. Da bei vielen Verfahren jedoch die Rechenzeit nahezu exponentiell mit der Zahl der Variablen ansteigt, ist es oftmals sinnvoll, eine Datenreduktion durchzuführen. Hierbei finden verschiedene Verfahren Verwendung, z.B.:
- Mittelwertbildung eines Intervalls:
Hier wird die Wellenzahlen-Skala des Infrarotspektrums in Intervalle unterteilt. Für jedes dieser Intervalle wird der jeweilige Mittelwert bestimmt. Die Mittelwerte stellen die Basisbereiche des datenreduzierten Spektrums dar.
- Hadamard-Transformation:
Bei dieser Methode wird mit dem Spektrum eine Hadamard-Transformation durchgeführt. Diese ist der Fourier-Transformation sehr ähnlich, wobei jedoch statt der Sinusfunktion eine Rechteckfunktion verwendet wird.
Von den so erhaltenen Hadamard-Koeffizienten wird eine bestimmte Anzahl an Koeffizienten gleich Null gesetzt. Die verbleibenden Koeffizienten werden in das Infrarotspektrum rücktransformiert. Je mehr Koeffizienten zuvor auf Null gesetzt worden sind, desto geringer ist die Auflösung des datenreduzierten Spektrums.
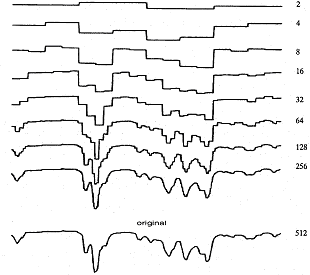
Vergleich des original IR-Spektrums (unten) mit den Datenreduzierten; Die Zahl des zurückbleibenden Hadamard-Koeffizienten bestimmt die Auflösung
- M. Razinger, M. Novic, Reduction of the Information Space for Data Collections, PCs for Chemists; Zupan, J. Ed.; Elsevier: Amsterdam 1990; pp 89-103;
- J. Zupan, Algorithms for Chemists, Wiley, New York 1989.
- M. Novic, J. Zupan, Chem. Inf. Comput. Sci. 1995, 35, 454-466.
© Prof. Dr. J. Gasteiger, Dr. Th. Engel, CCC Univ. Erlangen, Wed Jun 9 12:55:23 2004 GMT
 BMBF-Leitprojekt Vernetztes Studium - Chemie BMBF-Leitprojekt Vernetztes Studium - Chemie
|