Die Visualisierung biologischer Moleküle, die aus Hunderten
oder Tausenden von Atomen bestehen, ist mit Hilfe der bisher beschriebenen
Strukturmodelle (Draht-, Kugel-Stab- oder Kalottenmodell) nur bedingt
möglich. Zum einen werden diese Modelle bei mehr als Hundert
Atomen sehr schnell unübersichtlich und zum anderen sind die
erforderlichen Rechenleistungen zur interaktiven Visualisierung
solcher Moleküle recht hoch.
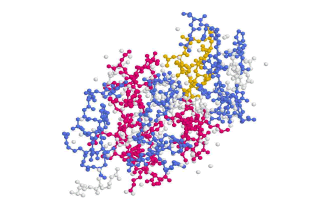
Lysozym dargestellt als Kugel-Stab-Modell in
Chime (links) und in VieweLite (rechts)
Zur Lösung dieser Problematik wurden einige vereinfachte Molekülmodelle
entwickelt, die in erster Linie zur Darstellung der Sekundärstrukturelemente
von Proteinen dienen. Dabei werden die Proteine meist als Bandmodell
visualisiert, wobei Sekundärstrukturen (Helices, Faltblätter)
besonders hervorgehoben und damit die Tertiärstruktur sichtbar
wird.
Ca-Trace-Modell
Dieses Modell repräsentiert ein Protein durch eine Linie
bzw. kleine röhrenförmige Gebilde (s. ViewerLite),
welche dem Backbone entlang, von a-Kohlenstoff
zu a-Kohlenstoff weiterführt.
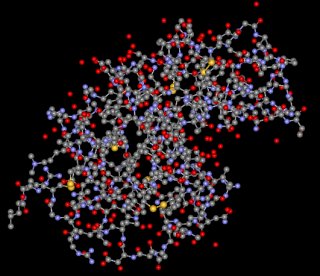
Lysozym dargestellt als Ca-Trace-Modell
in Chime (links) und in ViewerLite (rechts)
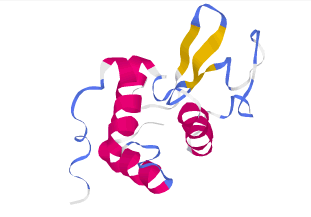
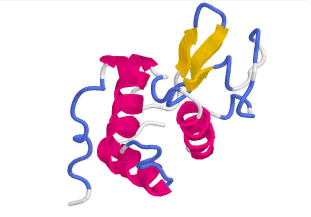
Bänder-Modell
Zur Visualisierung des Protein-Backbones hat sich das Bänder-
(Ribbon-) Modell etabliert. Ribbon-Modelle ähneln, wie
der Name schon sagt, in ihrem Aussehen flachen Bändern.
Die Oberseite dieser Bänder ist dabei parallel zur Peptidbindung
ausgerichtet. Das Bändermodell kann den Backbone als
Linien (strands), flaches oder als massives Band darstellen
(s. ViewerLite).
Lysozym dargestellt als Bändermodell
in Chime (links) und in ViewerLite (rechts)
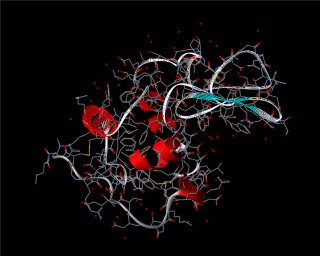
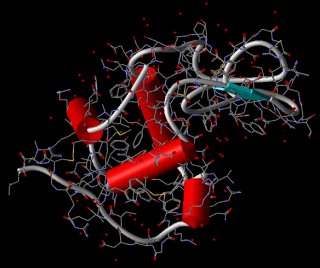
Schematische Darstellung
Eine etwas aufwendigere Darstellung des Backbones von Proteinen
erfolgt durch Zylinder, Pfeilstrukturen und
Tubes (Schläuche) (s. ViewerLite). Dabei werden
die Zylinder zur Kennzeichnung von a-Helices,
die Pfeilstrukturen, die in Richtung terminalen Kohlenstoff
zeigen, für b-Stränge
und Tubes für die restlichen Windungen (Coils und Turns)
verwendet.
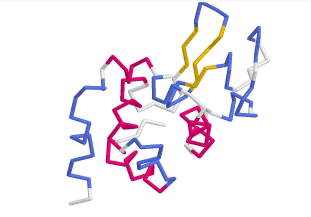
Lysozym als schematische Darstellung in
Chime (links: Cartoons) und in ViewerLite (rechts)