"Föllingsche Erkrankung"
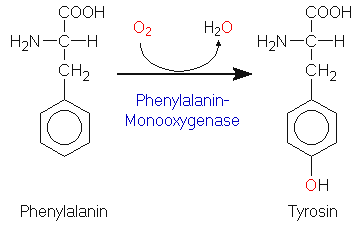
Tyrosin kann im menschlichen Organismus durch Oxidation aus Phenylalanin synthetisiert
werden. Die Reaktion wird durch das Enzym Phenylalanin-Monooxygenase
(Phenylalanin-Hydroxylase) katalysiert.
 |
Bei der Phenylketonurie fällt die Phenylalanin-Monooxygenase
vollständig aus oder ist in ihrer Funktion stark beeinträchtigt. Es handelt sich hierbei
um eine rezessiv vererbte Stoffwechselstörung (Häufigkeit: 1 : 10.000).
Die Aminosäure Tyrosin kann nicht mehr aus Phenylalanin synthetisiert
werden und ist daher essentiell.
Phenylalanin kann nicht über den üblichen Abbauweg: Tyrosin ®
Homogentisinsäure ® Fumarat + Acetoacetat abgebaut werden.
Es tritt daher die Phenylalanin-Transaminase in Aktion, wobei
Phenylpyruvat entsteht, das teilweise mit dem Harn ausgeschieden wird.
Phenylpyruvat ist jedoch neurotoxisch, es stört insbesondere das sich bei Kleinkindern erst
entwickelnde ZNS (vermutlich den Aufbau des Myelins). Daher muß die Phenylketonurie
baldmöglichst nach der Geburt (am 4. bis 6. Lebenstag) nachgewiesen werden um rechtzeitig
eine entsprechende Diät - phenylalaninarm und tyrosinreich
- geben zu können.