Einleitung
Da bisher keine Methoden existieren, die Verbindungseigenschaften direkt aus der molekularen Struktur vorhersagen, nutzen QSAR (quantitative structure activity relationship) und QSPR (quantitative structure property relationship) einen indirekten Ansatz. Dabei werden für eine Serie an Verbindungen zuerst numerische Deskriptoren berechnet, welche die Information der molekularen Struktur codieren. Diese Deskriptoren bzw. eine geeignete Teilmenge davon, werden dann in statistische Methoden oder künstliche neuronale Netz eingesetzt, um Modellezu entwickeln, die die gewünschte Eigenschaft oder Aktivität vorherzusagen.
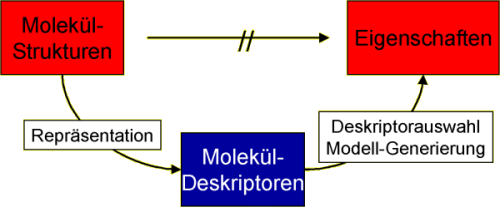
Moleküleigenschaften können nur indirekt über Deskriptoren vorhergesagt werden
© Prof. Dr. J. Gasteiger, Dr. Th. Engel, CCC Univ.
Erlangen, Wed Jun 9 12:55:24 2004 GMT
 BMBF-Leitprojekt
Vernetztes Studium - Chemie BMBF-Leitprojekt
Vernetztes Studium - Chemie
|